Medizin
Nr. 4 • April 2014
12
Serumkreatinins. Ab einer Protein-
ausscheidung > 300 mg in 24 Stunden
sind ACE-Hemmer indiziert. Bei allen
Sichelzellpatienten (v.a. HbSC-Patien-
ten) sollte jährlich eine Untersuchung
der Retina durchgeführt werden.
Einige Wochen vor einem langen
Flug sollte der Hb-Spiegel bei HbSC-
Patienten überprüft werden und evtl.
zur Ader gelassen werden.
Thalassämien
Als Thalassämien bezeichnet man
diejenigen Hämoglobinkrankheiten,
die durch eine reduzierte oder ganz
fehlende Synthese von α-, β-, γ- oder
δ-Globin-Ketten gekennzeichnet sind.
Fast allen ist eine Mikrozytose
gemein. Die Krankheitszeichen sind
proportional dem Ungleichgewicht
zwischen den in normaler Menge
gebildeten und den reduzierten Glo-
bin-Ketten.
β-Thalassämien
Bei den β-Thalassämien präzipitieren
die überschüssigen α-Ketten, die
keine Partner finden, um HbA (α 2 β 2 )
zu bilden. Diese Präzipitate schädigen
vor allem Normoblasten, was zur
einer ineffektiven Erythropoese führt.
Die Konsequenz ist eine gesteigerte
intestinale Eisenresorption, die auch
ohne Transfusion bei der schweren
β-Thalassaemia intermedia über die
Jahre zu einer chronischen Eisenüber-
ladung und daraus resultierenden
Organschäden führt [19].
Klinisch klassifiziert werden bei den
β-Thalassämien die β-Thalassaemia
minor, β-Thalassaemia major und
β-Thalassaemia intermedia. Indivi-
duen mit Thalassaemia minor sind
aymsptomatische Anlageträger, die
meisten keine Anämie haben. Thalas-
saemia major ist die schwerste Form,
bei der die Kinder im 1. Lebensjahr
transfusionspflichtig werden.
Sie ist zu unterscheiden von der Tha-
lassaemia intermedia, die jenseits des
1. Lebensjahres diagnostiziert wird.
Die Betroffenen sind nur unter bes-
timmten Bedingungen transfusions-
pflichtig. Die β-Thalassämie-Abgren-
zung „major“ und „intermedia“ ist in
der Praxis nicht leicht und wird meist
bereits vom pädiatrischen Hämatolo-
gen festgelegt. Da die Therapie der
β-Thalassaemia intermedia weniger
festen Vorgaben folgt wie die der Tha-
lassaemia major (nachzulesen in den
bereits erwähnten Leitfäden bzw.
Leitlinien) wird im Folgenden beson-
ders auf die β-Thalassaemia interme-
dia eingegangen.
Genetische Klassifizierung
Der Erbgang ist überwiegend autoso-
mal-rezessiv. Man spricht von einer
dominanten Form, wenn es bei eini-
gen seltenen Mutationen bei Hetero-
zygoten zu klinischen Manifestatio-
nen einer β-Thalassaemia intermedia
kommt. Auch bei Heterozygoten, die
zusätzlich eine Triplikation oder Qua-
druplikation einer α-Kette haben,
kommt es zur Intermedia.
Im Gegensatz zur α-Thalassämie, bei
der überwiegend Deletionen im α-
Globin-Gen vorherrschen, sind fast
alle β-Globin-Mutationen auf Punkt-
mutationen zurückzuführen (bisher
sind ca. 250 β-Globin-Mutationen
bekannt).
▶ β 0 -Mutation: keine β-Globin-Pro-
duktion
▶ β + -Mutation: erheblich verminder-
te β-Globin-Produktion
▶ β ++ -Mutation: gering verminderte
β-Globin-Produktion
In der Regel führt eine Homozygotie
für β 0 zu einer β-Thalassaemia major.
Die klinische Expression bei β-Tha-
lassämie ist aber sehr variabel. Ver-
schiedene modifizierende Faktoren
bestimmen den Schweregrad. Der
genetische Defekt erlaubt nicht die
Vorhersage des klinischen Schwere-
grads.
Diagnostik
Die für die Diagnose erforderlichen
Laboruntersuchungen sind einfach
und kostengünstig: Blutbild mit
RDW und Hämoglobinanalyse. Tha-
lassämie-Anlagenträger haben fast
immer einen Hb-Wert > 10 g/dl. Die
Hauptschritte sind in den Diagnostik-
Algorithmen (Abb. 2 und 3) darge-
stellt und in der aktuellen DGHO-
Leitlinie aufgeführt [5].
Die wichtigste Differenzialdiagnose
der Thalassämia minor ist der Eisen-
mangel. Allerdings ist im Gegensatz
zum Eisenmangel bei Thalassämia
minor die RDW und die Ferritin-Kon-
zentration meist normal. Diagnostisch
entscheidend ist die in der Hämoglo-
binanalyse erhöhte HbA 2 -Konzentra-
tion.
Thalassaemia major und intermedia
zeigen im Blutausstrich eine starke
Anisozytose (sehr hohe RDW).
Klinik
Patienten mit der schwer verlaufen-
den β-Thalassaemia major müssen
regelmässig transfundiert werden.
Infolgedessen entwickeln sie eine
Eisenüberladung und bedürfen bereits
ab dem Kleinkindalter einer täglichen
Eisenausschleusung [4, 5]. Die Betreu-
ung erfolgt nahezu ausschließlich in
den großen hämatologischen Zentren.
Kennzeichen eines schlecht betreuten
Patienten mit Thalassaemia major
oder intermedia ist die aufgrund der
gesteigerten ineffektiven Erythro-
poese ausgeweitete Maxilla-Spon-
giosa, die zu prominenten Jochbeinen
und einer Prognathie führt.
Etwa 1/5 der Thalassämien verlaufen
als Thalassaemia intermedia. Die
wesentlichen Krankheitszeichen sind
in Tab. 2 unter besonderer Berück-
sichtigung des Erwachsenenalters
aufgeführt.
Eine seltene, aber wichtige Komplika-
tion ist ein pulmonaler Hypertonus.
Bei einigen Patienten kann es zur
extramedullären Erythropoese mit
z.B. spangenartigen Tumoren um die
Wirbelsäule kommen. Asymptomati-
sche extramedulläre Tumore können
sich unter regelmäßigen Transfusio-
nen oder einer Hydroxycarbamid
Therapie zurückbilden. Bei sympto-
matischen Tumoren kann eine niedrig
dosierte Bestrahlung die beste
Behandlung sein.
Therapie
Die einzige kausale Behandlung ist die
allogene Stammzelltransplantation.
In den letzten Jahren konnte die Toxi-
zität der prätransplantären Kondi-
tionierung (Chemotherapie und
Bestrahlung) reduziert werden. Die
Verwendung haploidenter Spender
kann noch nicht standardmässig emp-
fohlen werden [14]. Die Hämoside-
rose mit all ihren Organendschäden
(endokrine Organe!) und das erhöhte
Abstoßungsrisiko durch die unzähli-
gen Transfusionen mindern den
Transplantationserfolg bei Erwachse-
nen. Daher sollten die Transplantatio-
nen vorzugsweise im Kindes- bzw.
Jugendalter erfolgen.
Sowohl für die Thalassämien als auch
für die Sichelzellerkrankung sind sog.
Genersatztherapien oder molekulare
Therapien, die den Switch vom fetalen
zum adulten Hämoglobin unterdrück-
en, mögliche, bisher jedoch nur
experimentell belegte Perspektiven
[23]. Patienten mit der Major-Form,
die keinen Stammzellspender haben,
müssen regelmäßig transfundiert
werden. Ziel ist es, die eigene Ery-
thropoese zu unterdrücken und ein
normales Wachstum zu ermöglichen,
um Knochendeformitäten zu vermei-
den.
Eine Therapie der Thalassaemia minor
ist nicht erforderlich. Schwangere
Anlageträgerinnen benötigen jedoch
Transfusionen, wenn der Hb-Wert
< 8 g/dl fällt und bei symptomatischer
Anämie.
Die Entscheidung zur regelmäßigen
Transfusion bei Patienten mit Tha-
lassaemia intermedia richtet sich
nach den im Therapie-Algorithmus
(Abb. 4) angegebenen Kriterien.
Patienten mit extramedullärer
Hämatopoese können entweder
transfundiert oder mit Hydroxycar-
bamid behandelt werden, sofern sie
keine neurologische Symptomatik
aufweisen.
Symptomatische Gallensteine sollten
entfernt werden (alle 3 Jahre Ultra-
schallkontrollen!). Ein schwieriges,
aber wichtiges Problem sind die
Ulcera cruris: die Therapie muss mul-
timodal erfolgen.
Die Eisenüberladung ist das Haupt-
problem bei allen β-Thalassämie-
Patienten. Bei der β-Thalassämie-
intermedia kommt es dazu auch ohne
Transfusion. Eine signifikante
Eisenüberladung kann bei einer Fer-
ritinkonzentration > 800 ng/ml
angenommen werden [20]. Die
Eisenüberladung der Leber und des
Herzens werden am besten mittels
MRT bestimmt und sollte bei Patien-
ten mit der Major-Form ab dem 12.
Lebensjahr jährlich, bei Patienten mit
der Intermedia-Form je nach Häufig-
keit von Transfusionen und Höhe des
Ferritinspiegels überwacht werden.
Die Hämochromatose des Herzens
mit ihren kardiopulmonalen Komp-
likationen ist die Haupttodesursache
älterer Thalassämie-Patienten. Die
Diagnostik und evtl. erforderliche
Dauertherapie bei kardiopulmonalen
Erkrankungen ist nicht Thalassämie-
spezifisch. Sie orientiert sich am
medizinischen Standard für die je-
weilige Komplikation.
Die Indikation zur Eisenchelation
wird gestellt, wenn eine oder mehrere
der folgenden Bedingungen erfüllt
sind:
▶ Ferritin-Konzentration von
> 800 μg/l (sekundäre Ferritinerhö-
hung bei akuten oder chronischen
Infekten, sowie bei Hepatitis C
beachten) [20].
▶ Lebereisenüberladung (methoden-
abhängig 3,2–7 mg/g (MRT) oder
4,5–9,6 mg/g Lebertrockengewicht)
[4, 19]. Die Autoren halten 7 mg/g
Lebereisen (im MRT bestimmt) für
eine gute Indikation zum Beginn
der Chelation.
▶ mehr als 10–15 Bluttransfusionen.
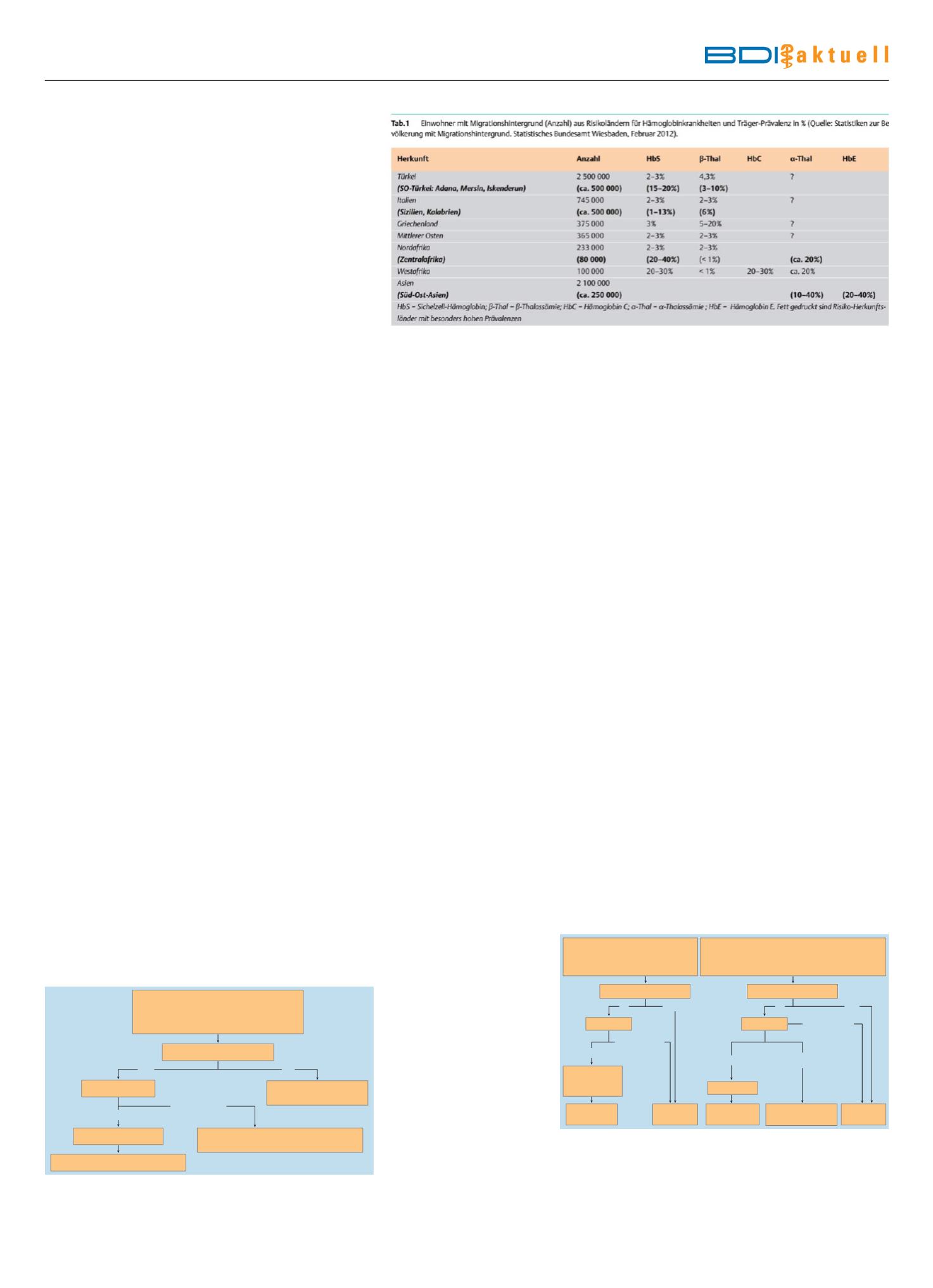
Kind 4–12 Monate
Hb<6 g/dl
MCV<70 fl, MCH<27 pg, RDW>15,5%
1
Ausgeprägte Symptome
2
Herkunft aus Endemieland?
Hb-Analyse
Hb-Analyse
Gen-Analyse
β-Thalassaemia
intermedia
Differenzial-
Diagnostik
5
β-Thalassaemia
minor+Eisenmangel
Untersuchung
beider Eltern, ggf.
Gen-Analyse
6
β-Thalassaemia
major
Differenzial-
Diagnostik
4
Herkunft aus Endemieland?
Erwachsener/Kind>12 Monate
Hb 6–11 g/dl
MCV<78 fl, MCH<27 pg, RDW>15,5%
1
Symptomatik variabel
3
nein
ja
Normalbefund oder
anormales Hb
HbF>10%
HbF<10%
HbA2>3%
nein
ja
HbF≥70%
Normalbefund oder
anormales Hb
Abb.2
Diagnostik-Algorithmus bei Verdacht auf Thalassaemia minor im Erwachsenenalter
[5]. 1 Orientierende Grenzwerte zur Einordnung der hypochromen, mikrozytären Anämie
bei diesen Patienten. 2 Eine mässige Blässe kann bestehen, kein Ikterus, keine Hepatosple-
nomegalie, keine Gewichtsabnahme; Anämie-Symptomatik bei gleichzeitigem Eisenman-
gel.
Erwachsene
Hb > 10 g/dl (auch Normalwert möglich)
MCV< 78 fl, MCH< 27 pg
1
keine ausgeprägten Symptome
2
Eisenmangel, RDW> 15,5%?
Hb-Analyse
β-Thalassaemia minor
Genanalyse bei klinischer Relevanz
Therapie
Blutbildkontrolle
Differenzial-Diagnostik
(z.B. α-Thalassämie, abnorme Hämoglobine)
nein
ja
HbA2>3,5%
Normalbefund oder
anormales Hb
Abb.3
Diagnostikalgorithmus bei manifester Thalassämie [5]:
1 Orientierende Grenzwerte zur Einordnung der hypochromen, mikrozytären Anämie bei
diesen Patienten
2 Blässe, Ikterus, Hepatosplenomegalie, Gedeihstörung
3 Breites Symptomspektrum zwischen Thalassaemia major und minor
4 Andere hereditäre hämolytische Anämie; andere hämatologische Erkrankung mit Anä-
mie und Organomegalie
5 Andere hereditäre oder erworbene, hypochrom mikrozytäre Anämien
6 Eine genetische Untersuchung ist bei Thalassaemia major prinzipiell entbehrlich. Sie
wird jedoch in den meisten Fällen durchgeführt.


