Basic HTML-Version




Medizin
Nr. 8/9 • August 2012
13
Anamnese
Eine 51-jährige Patientin stellte sich
mit seit 3 Wochen bestehenden mil-
den, uncharakteristischen, rechtsseiti-
gen Oberbauchbeschwerden in unserer
Ambulanz vor. Der Hausarzt hatte sie
zum Ausschluss einer Hernierung
zugewiesen. Die Krankengeschichte
war bis auf eine Unterbauchlaparosko-
pie bei Verdacht auf Endometriose und
eine konventionelle Ileozökalresektion
vor 7 Jahren unauffällig.
Körperlicher Untersuchungsbefund
Die allseits orientierte Patientin war in
befriedigendem Allgemeinzustand und
normalen Ernährungszustand (1,68 m,
65 kg, BMI 23). Das Integument war
unauffällig, enoral blande, Lungen
beidseits mit vesikulärem Atemge-
räusch, keine Atemnebengeräusche.
Herz rein und rhythmisch. Abdomen
unauffällig ohne Druckschmerz oder
Abwehrspannung. Ein neurologisches
Defizit war nicht zu eruieren. Die
erhobenen Kreislaufparameter waren
allesamt unauffällig und die Patientin
afebril (37,2 °C aurikulär).
Klinisch-chemische Untersuchung
Die laborchemischen Untersuchungen
waren unauffällig. Die bestimmten
Leberenzyme waren normwertig
(Bilirubin von 0,41 mg/dl: norm
0-1,0 mg/dl, AP von 66.1 U/l: norm
34 -105 U/l, GOT von 16,7 U/l: norm
< 33,0 U/l, GPT von 13 U/l: norm
< 32 U/l, GGT von 9,6 U/l: norm:
40 U/l).
Ergänzende Untersuchungen
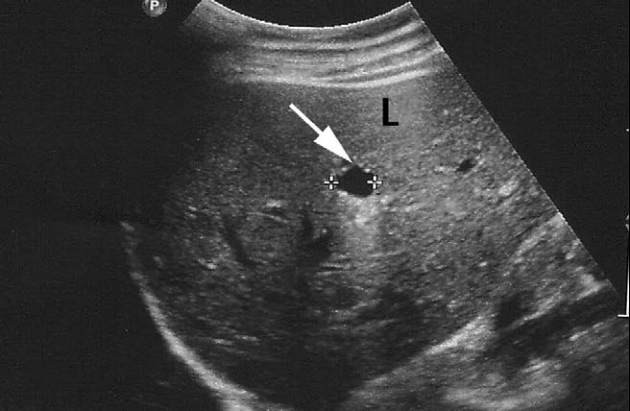
Bei der Sonographie des Abdomens
konnte die Gallenblase an der zu
erwartenden Stelle nicht dargestellt
werden. Der Ductus hepaticus com-
munis (DHC) war mit einem Durch-
messer vom 0,3 cm normal groß und
es zeigten sich multiple Zysten der
Leber. Der restliche Bauch war sono-
graphisch unauffällig (Abb. 1).
Zur Ergänzung der Diagnostik wurde
eine Magnetresonanztomographie
(MRT) der Leber durchgeführt. Hierbei
ließen sich weder die GB noch der D.
cysticus darstellen. Multiple Leberzys-
ten wurden in der MRT bestätigt,
andere Fehlbildungen bzw. Missbildun-
gen wurden nicht gesichtet
(Abb. 2, Abb. 3).
Therapie und Verlauf
Die Patientin wurde von uns ambulant
gesehen und war vor allen Dingen
besorgt und um eine kausale Abklä-
rung bedacht. Die Beschwerden an sich
waren auch subjektiv nur unwesent-
lich einschränkend, die Ausprägung
gering. Unter dem Verdacht einer Gas-
tritis wurde nach Ausschluss einer
akuten interventionsbedürftigen
Pathologie eine Medikation mit Proto-
nenpumpeninhibitoren empfohlen. Bei
Persistieren der Symptomatik wurde
ihr ergänzend zu einer Gastroösopha-
goskopie geraten.
Die Gallenblasenagenesie betreffend
wurde sie ausführlich über die in die-
sem Fall harmlose Diagnose aufgeklärt.
Diskussion
Die Gallenblasenagenesie ist eine sehr
seltene kongenitale Fehlbildung der
Gallenblase einschließlich des Ductus
cysticus bei sonst normalem intra-
und extrahepatischem Gallengangssys-
tem. Der erste Fall einer Gallenblasena-
genesie wurde schon im 17. Jahrhun-
dert von Lemery und Bergman
beschrieben. Seitdem wurden knapp
400 Fälle in der medizinischen Litera-
tur publiziert. Pathogenetisch soll eine
embryonale Hemmungsfehlbildung
des hepatischen Divertikulums des
Darmrohrs in der 3. Entwicklungs-
woche vorliegen [3]. Einer Arbeit von
Mannl [8] zufolge liegt die Rate von
Gallenblasenagenesien bei Kindern von
in der Schwangerschaft mit Thalido-
mid behandelten Mutter bei ca. 10 %.
Die Prävalenz in der Allgemeinbevöl-
kerung wird jedoch nur auf 0,007–
0,06 % geschätzt. Hierbei ist die klini-
sche Inzidenz mit ca. 0,007–0,027 %
deutlich geringer als die Inzidenz bei
Autopsien (0,04-0,13 %). Frauen wer-
den mit einem Verhältnis von 3:1 häu-
figer als Männer betroffen [9].
Im Kindesalter tritt die Gallenblasen-
agenesie in Kombination mit Fehlbil-
dungen des urogenitalen, gastrointesti-
nalen und kardiovaskulären Systems
als ein Bestandteil von Syndromen
gehäuft auf [12]. Eine genetische Prä-
disposition wird nach dem Auftreten
in über zwei Generationen diskutiert.
Das klinische Bild der Gallenblasen-
agenesie ist relativ bunt. Bennion et al.
[2] unterschieden nach einer Analyse
von über 380 Fällen drei Verlaufsfor-
men bzw. Gruppen. Gruppe I (ca. 15 %)
ist durch multiple fetale Anomalien
meistens im Kindesalter gekennzeich-
net. Der Verlauf endet fast immer letal.
Gruppe II (ca. 35 %) umfasst asympto-
matische Patienten, während die
Gruppe III (ca. 50 %) symptomatische
Patienten beinhaltet.
Symptome beginnen überwiegend
zwischen der 4. und 5. Lebensdekade
mit rezidivierenden rechtsseitigen
Oberbauchschmerzen mit Koliken,
Cholangitiden und Ikterus. Diese
Beschwerden sind ebenso typisch für
andere Pathologien der Gallenblase
und des Gallengangssystems, sodass
eine spezifische klinische Zuordnung
zu der Gallenblasenagenesie nicht
möglich ist [7]. Laut Dickinson et al. [4]
wird eine Erweiterung des Ductus
hepaticus communis in bis zu 50 % der
Fälle beobachtet. Hierher findet sich
eine Choledocholithiasis in ca. 27 % der
Fälle.
Die Diagnostik der Gallenblasenagene-
sie kann wie in dem von Vijay et al.
[13] beschriebenen Fall eine Heraus-
forderung sein. In erster Linie spielen
laborchemische Untersuchungen ins-
besondere die Bestimmung der Leber-
transaminasen und des Bilirubinwer-
tes eine große Rolle. Die apparative
Dia-gnostik erfolgt überwiegend sono-
graphisch. Hierbei orientiert man sich
anhand der WES-Trias (W: Gallenbla-
senwand, E: Steinecho, S: Schallschat-
ten) nach MacDonald et al. [6]. Da die
WES-Trias bei Schrumpf-Gallenblasen
unzuverlässig zu sein scheint und die
Sonographie vom Untersucher abhän-
gig ist, ist der Ultraschall keine ausrei-
chende Diagnostik der Gallenblasena-
genesie. Hier ist die Magnetresonanz-
Tomographie (MRT) mit Darstellung
des Gallengangsystem (MRCP) eine
hervorragende nicht invasive diagnos-
tische Ergänzung [10, 11].
In dem 20. Jahrhundert wurde die
Diagnose der Gallenblasenagenesie
meist intraoperativ gestellt [1]. Hierbei
dürfte laut Frey [5] eine Gallenblasen-
agenesie erst diagnostiziert werden,
wenn eine ektope Gallenblase im Rah-
men einer Laparotomie ausgeschlossen
wurde. Dies ist heutzutage wegen des
verbreiteten Einsatzes der MRT obso-
let. Aus gleichem Grund wird die
endoskopische retrograde Cholangio-
Pankreatikographie (ERCP) lediglich
bei einer Cholestase zur Abklärung
empfohlen. Hierbei lässt sich in den
Gallenblasenagenesie – eine seltene
kongenitale Fehlbildung
Kasuistik
Die Gallenblasenagenesie ist eine sehr seltene kongenitale Fehlbildung der Gallenblase. Die Gallenblase
ist dabei inklusive des Ductus cysticus (Gallenblasengangs) bei sonst normalem intra- und extrahepa-
tischem Gangsystem nicht angelegt. Das klinische Spektrum dieser Fehlbildung reicht von kompletter
Symptomlosigkeit bis zu Oberbauchkoliken und Gelbsucht. Die Behandlungsindikation richtet sich nach
den Beschwerden, wobei die Diagnosestellung durchaus sehr herausfördernd sein kann.
Abb. 1
Oberbauchsonographie ohne Nachweis einer Gallenblase an der typischen Stelle.
Abb. 2
Magnetresonanztomographie des Oberbauchs in T2-Wichtung ohne Nachweis
einer Gallenblase.
Abb. 3
Magnetresonanz-Cholangiopankreatikographie (MRCP) mit fehlender Gallanblase.
Auch der Ductus cysticus ist nicht darstellbar.